Amyloid PET Centiloid Pipeline
Below is presented a pipeline for CL analysis of static amyloid brain PET scan using [18F] florbetaben radiotracer.
The scan consists of four 5-minutes frames which are first aligned and then processed for CL.
First, import all necessary Python packages
from pathlib import Path
import numpy as np
from matplotlib import pyplot as plt
from niftypet import nimpa
import spm12
import amypet
from amypet import backend_centiloid as centiloid
from amypet import params as Cnt
import logging
logging.basicConfig(level=logging.INFO)
Set up the input and output
# > ignore derived DICOM files
ignore_derived = False
# > uptake ratio (UR) window def (unfortunately, aka SUVr)
ur_win_def = [5400,6600]
# > type of amyloid radiopharmaceutical, here [18F]florbetaben
tracer = 'fbb'
# > input PET folder
input_fldr = Path('/home/pawel/data/PNHS/FBB1_STAT')
# > output path
outpath = input_fldr.parent/('amypet_output_'+input_fldr.name)
Please note that not infrequently DICOM fields have wrong tracer information recorded, e.g., FDG instead of an amyloid tracer name. Hence it is always safer to define this prior to the analysis as it is done above.
It is also important to note that for a proper functioning of this pipeline there are DICOM fields which cannot be missing about the PET acquisition - see below for details.
Explore, identify DICOM files and convert to NIfTI
Please note, that in order to obtain the relevant information about PET acquisition, key DICOM fields have to be present. Among others (such as image orientation and size), these are:
- study time and date
- series time
- acquisition time
- frame duration time
- time of tracer administration (start)
- PET tracer name (not always accurate or present; can be overwritten by specifying it upfront when running AmyPET).
#------------------------------
# > structural/T1w image
ft1w = amypet.get_t1(input_fldr, Cnt)
if ft1w is None:
raise ValueError('Could not find the necessary T1w DICOM or NIfTI images')
#------------------------------
#------------------------------
# > processed the PET input data and classify it (e.g., automatically identify UR frames)
indat = amypet.explore_indicom(
input_fldr,
Cnt,
tracer=tracer,
find_ur=True,
ur_win_def=ur_win_def,
outpath=outpath/'DICOMs')
# > convert to NIfTIs
niidat = amypet.convert2nii(indat, use_stored=True)
#------------------------------
Align the dynamic/static frames
This does not have to be executed if only one PET frame is available. The alignment can be done with SPM or DIPY registration tools.
aligned = amypet.align(
niidat,
Cnt,
reg_tool='spm',
ur_fwhm=4.5,
use_stored=True)
CL quantification
# > optional CSV file output for CL results
fcsv = input_fldr.parent/(input_fldr.name+'_AmyPET_CL.csv')
out_cl = centiloid.run(
aligned['ur']['fur'],
ft1w,
Cnt,
stage='f',
voxsz=1,
bias_corr=True,
tracer=tracer,
outpath=outpath/'CL1',
use_stored=True,
#fcsv=fcsv
)
Exploring the output
The output dictionary out_cl contains the following outputs:
out_cl['fmri']: file path to the original input T1w MRI image.
out_cl['n4']: output of the bias field correction for the T1w MRI image.
out_cl['mric']: output of the centre-of-mass correction for the MRI image, including output file path as well as relative and absolute translation parameters.
out_cl['petc']: output of the centre-of-mass correction for PET image, including output file path as well as relative and absolute translation parameters.
Please note that all the output for centre-of-mass corrections are stored in subfolder centre-of-mass inside the output folder out_cl['opth'].
out_cl['fpet']: the path to the CL input PET after alignment which includes centre of mass adjustment to facilitate more robust SPM registration/alignment.
out_cl['reg1']: output of the affine MRI registration to the MNI space template; output includes the path to the registered MRI image, path to the affine transformation file, and the affine transformation array itself.
out_cl['reg2']: output of the affine PET registration to the above registered MRI image; output includes the path to the registered PET image, path to the affine transformation file, and the affine transformation array itself.
Please note that all that all the affine registration output is stored in subfolder registration in the output folder out_cl['opth'].
out_cl['norm']: output of all the SPM normalisation/segmentation, including paths to the parameters, inverse and forward transfromations as well as the grey (c1) and white (c2) matter segmentation probability masks.
out_cl['fnorm']: is the list of all the paths of spatially normalised files, including the MRI, PET and grey/white matter segmentation probability masks.
out_cl['avgvoi']: is the output dicionary of average volume of interest (VOI) values for the Centiloid masks of the cerebellum grey matter (cg), the whole cerebellum (wc), the whole cerebellum + brain stem (wcb), the pons (pns) and the cortex (ctx).
out_cl['ur']: are the uptake ratio values (unfortunately also known as SUVr) for the cerebellum grey matter (cg), the whole cerebellum (wc), the whole cerebellum + brain stem (wcb), the pons (pns) reference regions.
out_cl['ur_pib_calc']: these are the [C11]PiB converted values to the above.
out_cl['ur_pib_calc_transf']: are the saved linear transformation parameters.
Main output:
out_cl['cl']: the Centiloid (CL) values for each reference region, that is the cerebellum grey matter (cg), the whole cerebellum (wc), the whole cerebellum + brain stem (wcb), the pons (pns).
out_cl['fcsv']: the path to the CSV file with all the outputs.
out_cl['fqc']: the file path to the QC image showing all the sampling masks superimposed on the PET image in transaxial and sagittal views together with printed all the CL and UR values.
Image Output:
The key image output in NIfTI format for the CL converted images suing all the available reference regions (the cerebellum grey matter, the whole cerebellum, the whole cerebellum + brain stem , the pons) as well as the corresponding uptake ratio (UR) images (aka SUVr) are contained in the results subfolder folder.
The QC image for the Centiloid pipeline can be viewed as follows:
from PIL import Image
fig,axs = plt.subplots(1,1, figsize=(16, 12))
# > show the QC image in PNG format for the the Centiloid pipeline
axs.imshow( np.asarray(Image.open(out_cl['fqc'])) )
axs.axes.get_xaxis().set_ticks([])
axs.axes.get_yaxis().set_ticks([])

Quality Control Checks
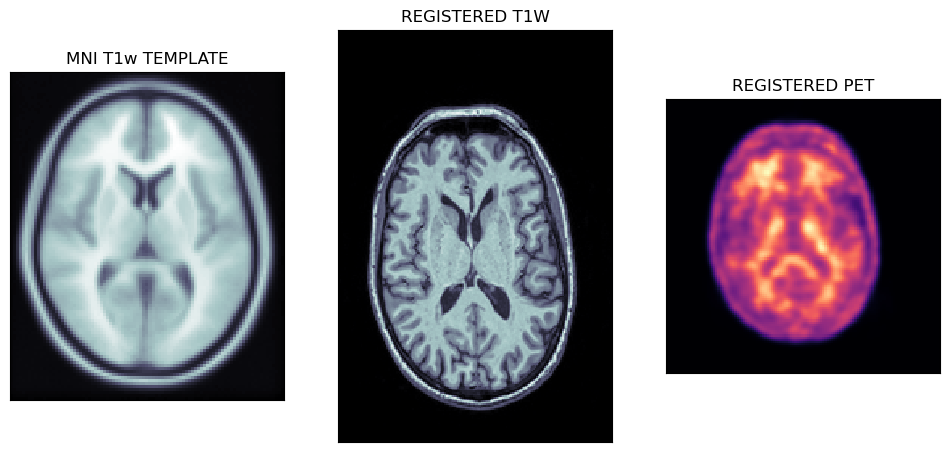
Viewing affine registered MRI and PET images
The first output of the CL pipeline are the registrations of T1w MRI image to the MNI template followed by the registration of PET to the MNI registered MRI image.
tmpl = nimpa.getnii(spm12.spm_dir()+'/canonical/avg152T1.nii')
t1wa = nimpa.getnii(out_cl['reg1']['freg'])
peta = nimpa.getnii(out_cl['reg2']['freg'])
fig,axs = plt.subplots(1,3, figsize=(12, 8))
axs[0].matshow(tmpl[50,...], cmap='bone')
axs[1].matshow(t1wa[95,...], cmap='bone')
axs[2].matshow(peta[40,...], cmap='magma')
# > remove ticks
for i in range(3):
axs[i].axes.get_xaxis().set_ticks([])
axs[i].axes.get_yaxis().set_ticks([])
axs[0].set_title('MNI T1w TEMPLATE')
axs[1].set_title('REGISTERED T1W')
axs[2].set_title('REGISTERED PET')
The plots below show the MNI template with the registered MRI and PET images.
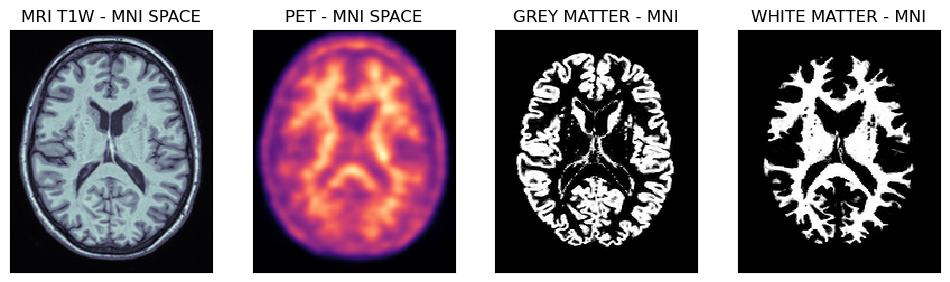
Viewing spatially normalised masks and MRI/PET images
Below are shown PET and MRI images spatially normalised to the MNI space. Note, that the voxel size is 1 mm isotropically, hence the probability masks for grey and white matter regions are well defined.
mrin = nimpa.getnii(out_cl['fnorm'][0])
petn = nimpa.getnii(out_cl['fnorm'][1])
gmn = nimpa.getnii(out_cl['fnorm'][2])
wmn = nimpa.getnii(out_cl['fnorm'][3])
fig,axs = plt.subplots(1,4, figsize=(12, 8))
axs[0].matshow(mrin[95,...], cmap='bone')
axs[1].matshow(petn[95,...], cmap='magma')
axs[2].matshow(gmn[95,...], cmap='gray')
axs[3].matshow(wmn[95,...], cmap='gray')
# > remove ticks
for i in range(4):
axs[i].axes.get_xaxis().set_ticks([])
axs[i].axes.get_yaxis().set_ticks([])
axs[0].set_title('MRI T1W - MNI SPACE')
axs[1].set_title('PET - MNI SPACE')
axs[2].set_title('GREY MATTER - MNI')
axs[3].set_title('WHITE MATTER - MNI')
fig,axs = plt.subplots(1,2, figsize=(10, 6))
axs[0].matshow(petn[95,...], cmap='magma')
axs[0].imshow(gmn[95,...], cmap='gray', alpha=0.3)
axs[1].matshow(petn[95,...], cmap='magma')
axs[1].imshow(wmn[95,...], cmap='gray', alpha=0.3)
# > remove ticks
for i in range(2):
axs[i].axes.get_xaxis().set_ticks([])
axs[i].axes.get_yaxis().set_ticks([])
axs[0].set_title('GM MASK IN PET')
axs[1].set_title('WM MASK IN PET')
Transaxial slices of the MRI and PET images in the MNI space together with the grey and white matter probability masks. Further below are shown the grey matter and white matter masks superimposed on the PET image.